功能: catalytic activity:Hydrolysis of terminal non-reducing beta-D-galactose residues in beta-D-galactosides.,disease:Defects in GLB1 are the cause of GM1-gangliosidosis type 1 (GM1G1) [MIM:230500]; also known as infantile GM1-gangliosidosis. GM1-gangliosidosis is an autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1G1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life.,disease:Defects in GLB1 are the cause of GM1-gangliosidosis type 2 (GM1G2) [MIM:230600]; also known as late infantile/juvenile GM1-gangliosidosis. GM1G2 is characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive.,disease:Defects in GLB1 are the cause of GM1-gangliosidosis type 3 (GM1G3) [MIM:230650]; also known as adult or chronic GM1-gangliosidosis. GM1G3 is characterized by a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive.,disease:Defects in GLB1 are the cause of mucopolysaccharidosis type 4B (MPS4B) [MIM:253010]; also known as Morquio syndrome B. MPS4B is a form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life.,function:Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans.,function:Isoform 2 has no beta-galactosidase catalytic activity, but plays functional roles in the formation of extracellular elastic fibers (elastogenesis) and in the development of connective tissue. Seems to be identical to the elastin-binding protein (EBP), a major component of the non-integrin cell surface receptor expressed on fibroblasts, smooth muscle cells, chondroblasts, leukocytes, and certain cancer cell types. In elastin producing cells, associates with tropoelastin intracellularly and functions as a recycling molecular chaperone which facilitates the secretions of tropoelastin and its assembly into elastic fibers.,online information:Beta-galactosidase entry,similarity:Belongs to the glycosyl hydrolase 35 family.,subcellular location:Localized to the perinuclear area of the cytoplasm but not to lysosomes.,
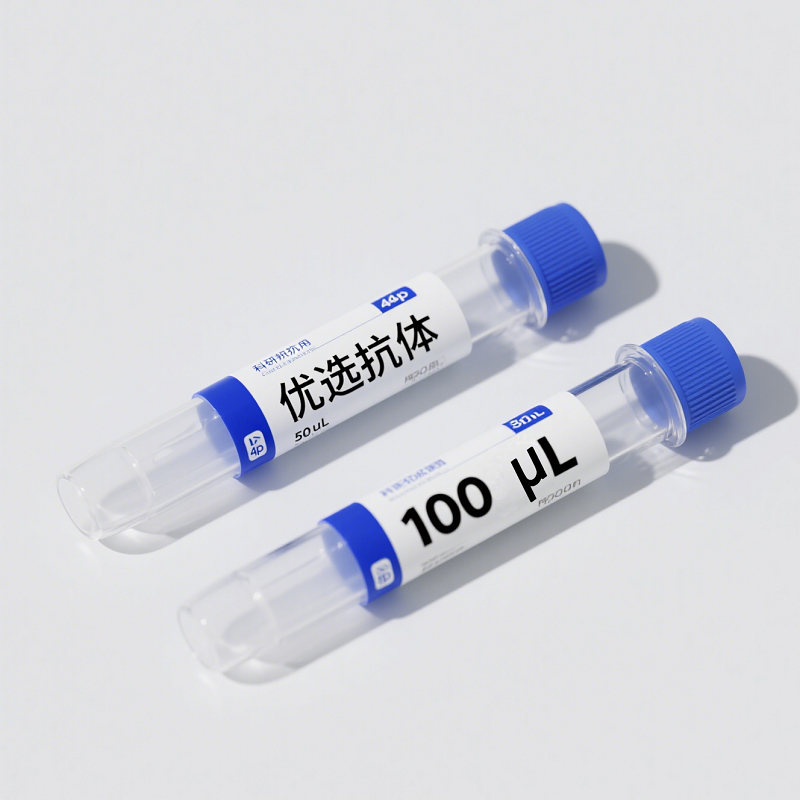
相关产品: RS0001,RS0002,YM3028,YM3029
细胞定位: [Isoform 1]: Lysosome .; [Isoform 2]: Cytoplasm, perinuclear region . Localized to the perinuclear area of the cytoplasm but not to lysosomes. .
组织表达: Detected in placenta (at protein level) (PubMed:8383699). Detected in fibroblasts and testis (PubMed:2511208).
功能: catalytic activity:Hydrolysis of terminal non-reducing beta-D-galactose residues in beta-D-galactosides.,disease:Defects in GLB1 are the cause of GM1-gangliosidosis type 1 (GM1G1) [MIM:230500]; also known as infantile GM1-gangliosidosis. GM1-gangliosidosis is an autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1G1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life.,disease:Defects in GLB1 are the cause of GM1-gangliosidosis type 2 (GM1G2) [MIM:230600]; also known as late infantile/juvenile GM1-gangliosidosis. GM1G2 is characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive.,disease:Defects in GLB1 are the cause of GM1-gangliosidosis type 3 (GM1G3) [MIM:230650]; also known as adult or chronic GM1-gangliosidosis. GM1G3 is characterized by a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive.,disease:Defects in GLB1 are the cause of mucopolysaccharidosis type 4B (MPS4B) [MIM:253010]; also known as Morquio syndrome B. MPS4B is a form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life.,function:Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans.,function:Isoform 2 has no beta-galactosidase catalytic activity, but plays functional roles in the formation of extracellular elastic fibers (elastogenesis) and in the development of connective tissue. Seems to be identical to the elastin-binding protein (EBP), a major component of the non-integrin cell surface receptor expressed on fibroblasts, smooth muscle cells, chondroblasts, leukocytes, and certain cancer cell types. In elastin producing cells, associates with tropoelastin intracellularly and functions as a recycling molecular chaperone which facilitates the secretions of tropoelastin and its assembly into elastic fibers.,online information:Beta-galactosidase entry,similarity:Belongs to the glycosyl hydrolase 35 family.,subcellular location:Localized to the perinuclear area of the cytoplasm but not to lysosomes.,
相关产品: RS0001,RS0002,YM3028,YM3029
细胞定位: [Isoform 1]: Lysosome .; [Isoform 2]: Cytoplasm, perinuclear region . Localized to the perinuclear area of the cytoplasm but not to lysosomes. .
组织表达: Detected in placenta (at protein level) (PubMed:8383699). Detected in fibroblasts and testis (PubMed:2511208).

 在线咨询
在线咨询








 湘公网安备
湘公网安备